Gaussian
This tutorial demonstrates how to generate Gaussian input files for Osmocene using Avogadro 1.2.0 and prepare a geometry optimization job with DFT (B3LYP/LANL2DZ). The process includes molecule building, editing, and input generation via the Avogadro graphical user interface.
Getting Started
1. Install and Launch Avogadro
- Download Avogadro from the official website.
- Run the installer and complete the setup.
- Launch Avogadro.
Note: Avogadro provides several essential tools such as Draw, Navigation, Manipulation, and Selection to build and edit molecules. These tools appear in the toolbar by default; if they are not visible, enable them by going to
Settings → Toolbarsand checking the “Tools” option. For detailed descriptions and usage instructions, see the Avogadro tools documentation.
2. Building the Osmocene Molecule
- Navigate to
Build → Insert → Fragment. -
Search for
cyclopentadienyland insert the fragment into the workspace.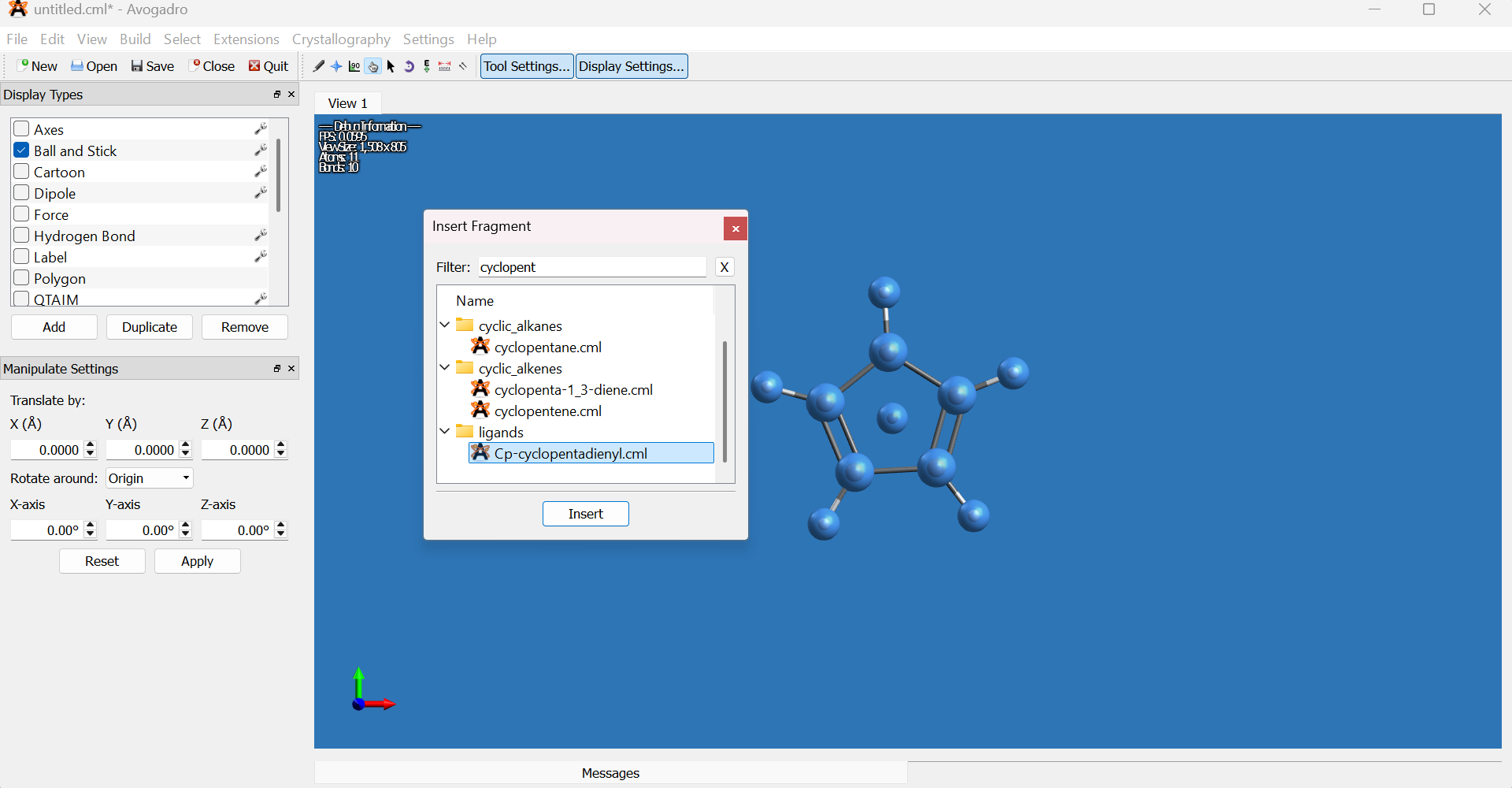
- Copy the fragment using
Edit → Copy(orCtrl+C). - Paste the copied fragment with
Edit → Paste(orCtrl+V). The new fragment will overlap the original and remain selected. -
Translate one cyclopentadienyl ring approximately 3.0 Å along the Z-axis using the Translate option in the Manipulate Settings panel.
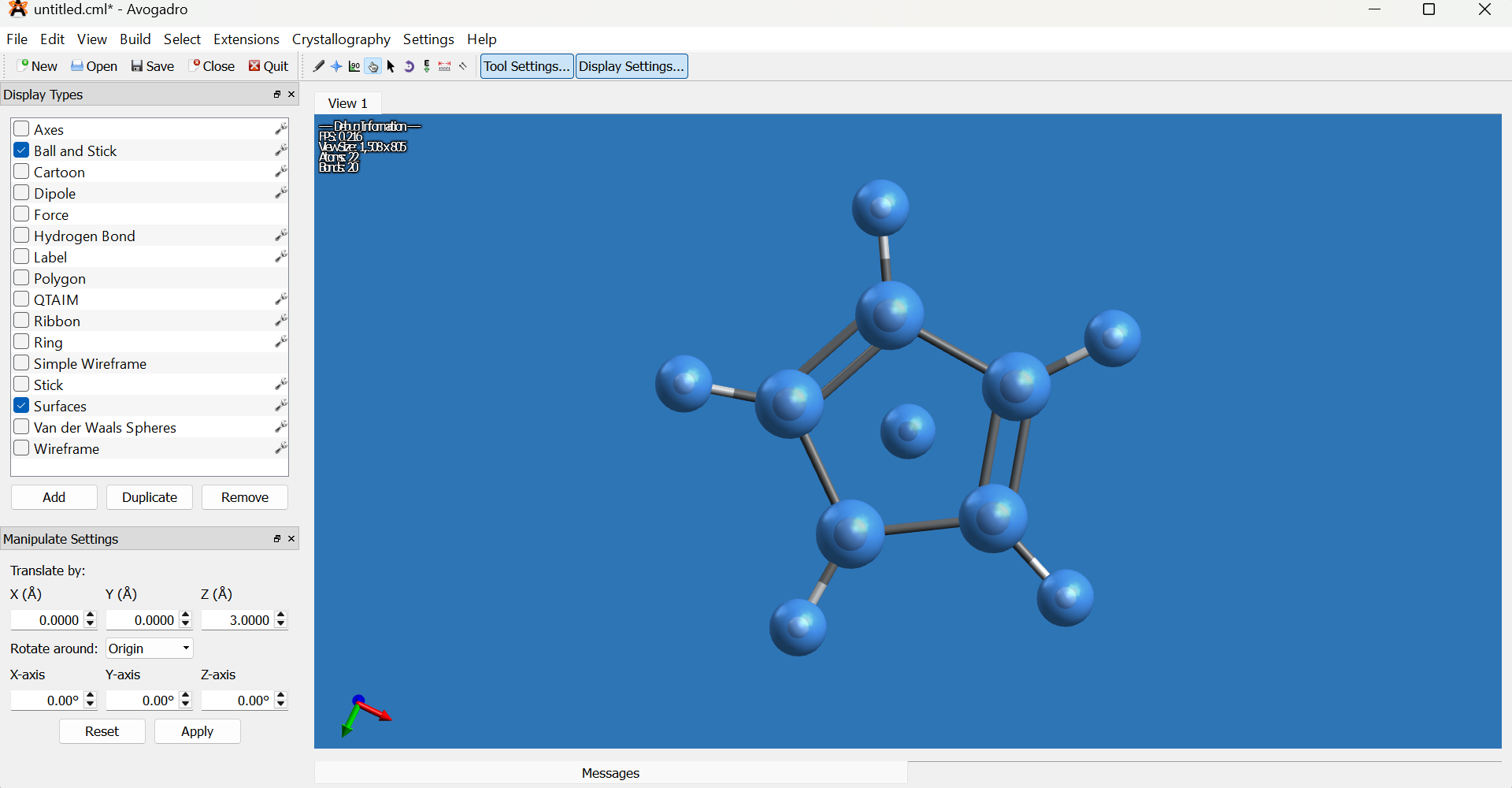
- If the Manipulate Settings panel does not appear, enable it via
Settings → Toolbars → Manipulate Settings. - Deselect all fragments (
Select → Select NoneorCtrl+Shift+A). -
Switch to the Draw tool, set the element to osmium (Os) from the element selector on the left bottom, then click one of the dummy atom in the middle of the rings to replace it with osmium.
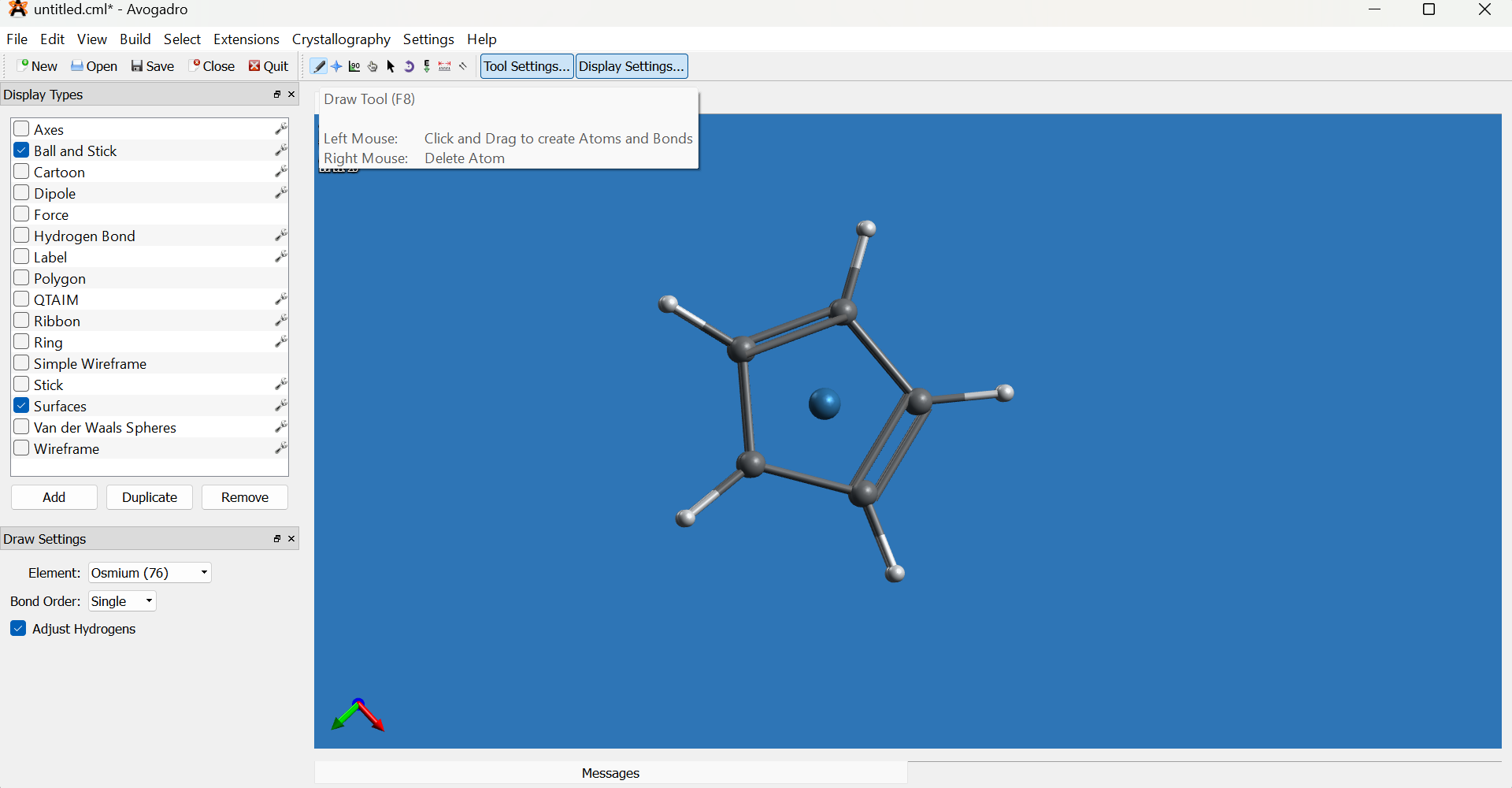
-
Select the Manipulation tool (hand icon) and rotate the molecule to view it from the side.
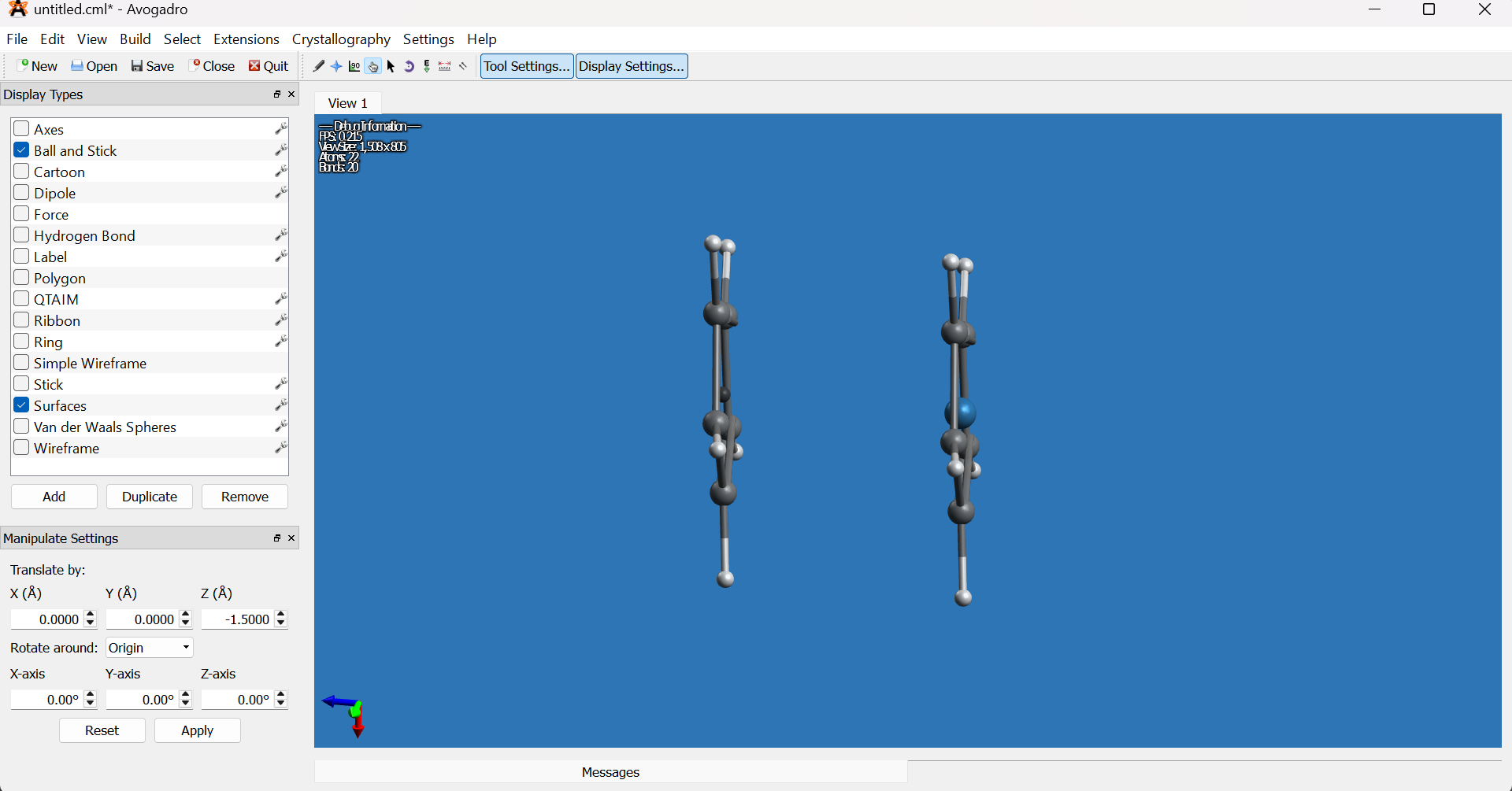
- Using the Selection tool (arrow icon), select the remaining dummy atom and delete it.
-
Select the osmium atom, then use the Manipulation tool to center it between the two cyclopentadienyl rings along the Z axis.
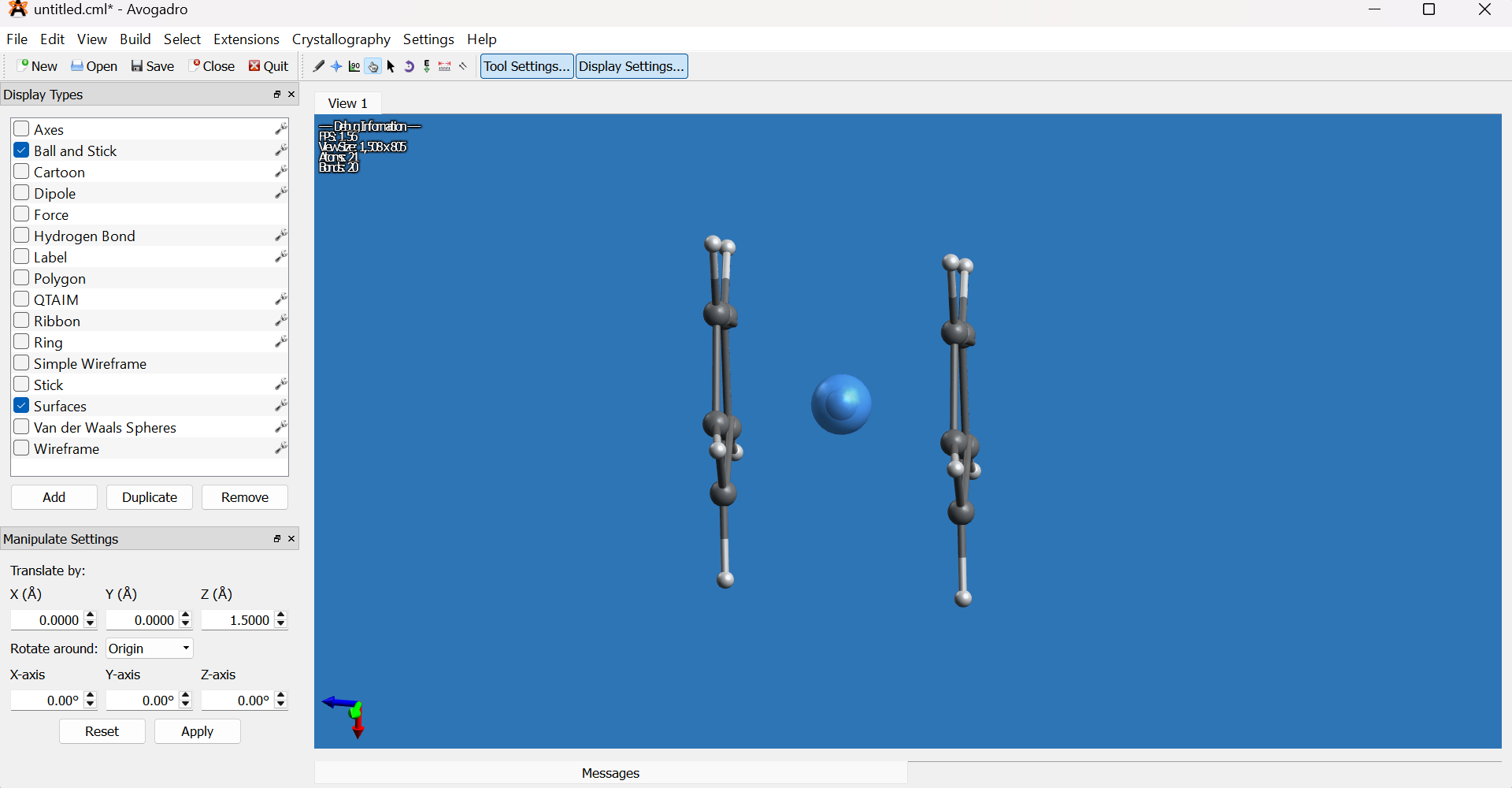
-
Deselect all (
Select → Select NoneorCtrl+Shift+A) and rotate to inspect the molecule from your preferred angle.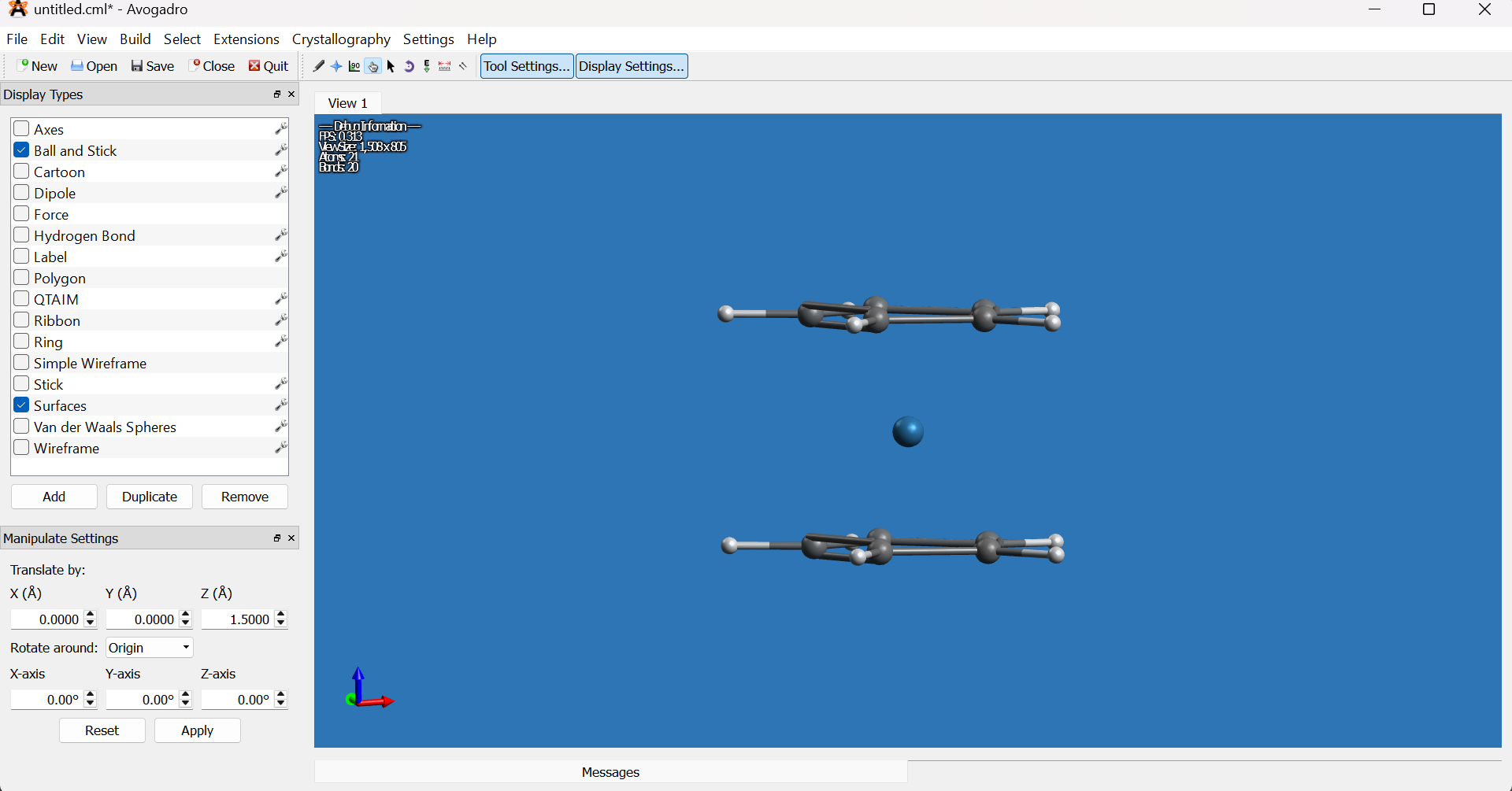
3. Generating Gaussian Input
- Go to
Extensions → Gaussian. - In the Gaussian input dialog:
- Set the Title to the molecule name (e.g., osmocene).
- Set Calculation to Geometry Optimization.
- Set Method to DFT (B3LYP).
- Set Basis Set to LANL2DZ.
- Adjust any additional keywords or options as necessary.
-
Click Generate to produce the Gaussian input file.
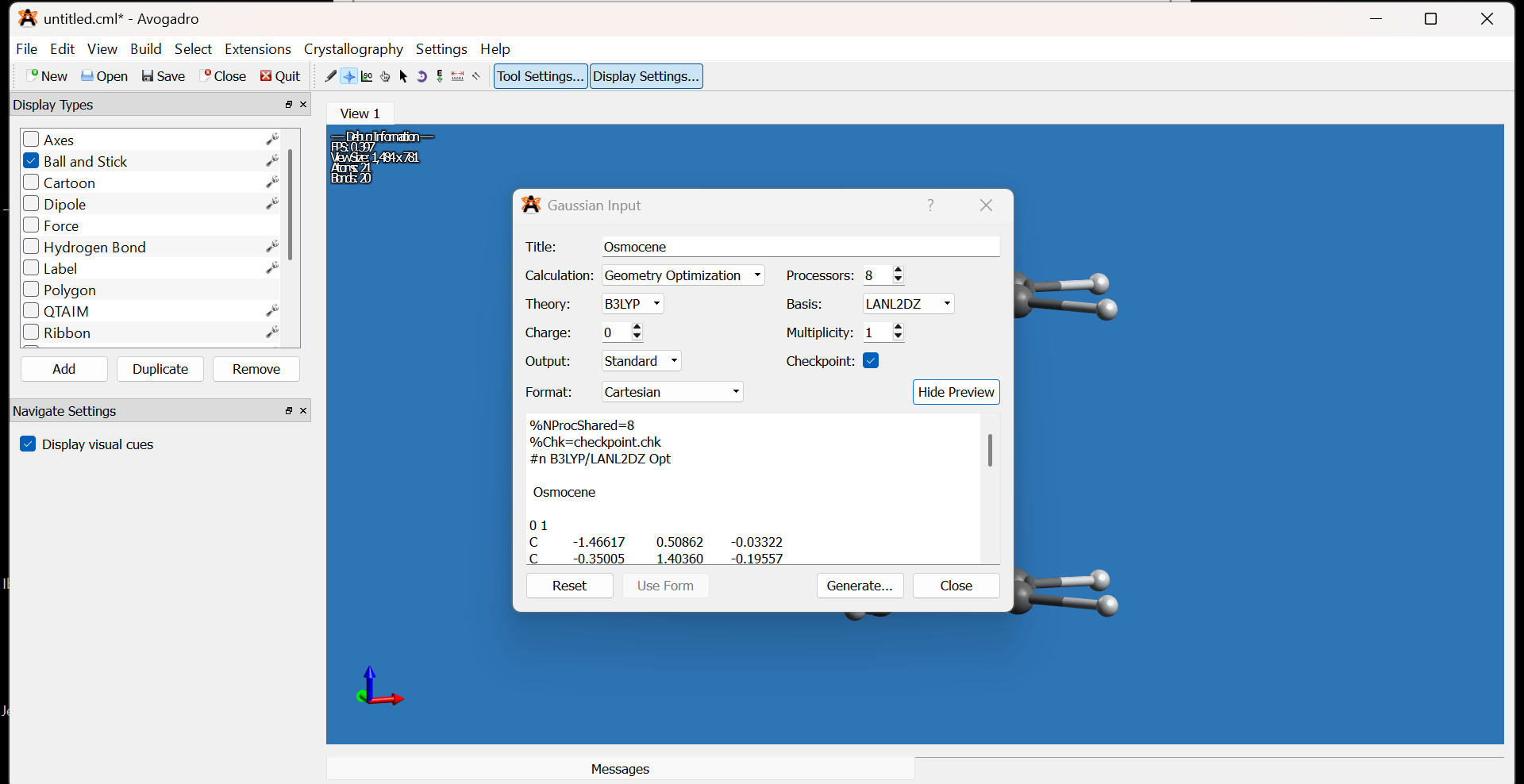
Note: There is no strict rule for choosing the DFT method or basis set. Refer to benchmarking studies and literature for your particular system to improve reproducibility.
Useful references:
4. Submitting Gaussian and ORCA Jobs on Clusters
- Connect to the HPC clusters using SSH with X11 forwarding enabled:
ssh -X username@ip-address
Available clusters and their IP addresses:
- IPC:
10.44.82.80 - Tsunami2:
10.44.82.85 -
Tsunami3:
10.44.82.86 - Gaussian job submission:
- On IPC cluster, run:
g16c -intel -time 96:0:0 filename.comor
g09d -intel -time 96:0:0 filename.com(Maximum runtime: 96 hours)
- On Tsunami2 and Tsunami3 clusters, run:
g16c filename.gjfor
g09d filename.gjf - ORCA job submission:
- On IPC cluster, execute:
qorca -intel -v4 -time 96:0:0 filename.inp filename.out - On Tsunami2 and Tsunami3 clusters, use:
qorca5 filename.inpor
qorca6 filename.inp
Note: Adjust the filenames (
filename.com,filename.gjf,filename.inp,filename.out) to match your input and output file names.
Further Resources
- For detailed Avogadro documentation, visit https://avogadro.cc/docs/.
- For detailed Gaussian documentation, visit https://gaussian.com/videos/.
- For detailed Orca documentation, visit https://www.faccts.de/docs/orca/6.0/manual/.